
The world of medical device software is constantly evolving and it is critical to understand the regulations to ensure your device is safe and effective. While technology has brought benefits to patient care, it has also introduced new risks and safety concerns for the patients who we are trying to help. With new technologies like health-related Artificial Intelligence (AI) we have the potential to reduce medical errors and provide accurate diagnosis. However, with all new technologies unintended consequences almost always occur. Medical device software differs from other software products in that your code has the potential to harm or kill someone. While providing radiation therapy to cancer patients, the Therac-25 software malfunctioned and killed at least six people.
Throughout my career, I have led the development of several medical devices including:
- Patient Controlled Analgesic Pumps
- Blood Glucose Monitors
- Electronic Medical Record (EMR) Systems
- Blood Analyzers
I have learned what does and doesn’t work over my tenure. I want to cover four things that everyone should know who is involved with developing medical device software.
Develop and test your safety controls
Over my 15-year career developing medical device software I have seen good designs and some that cause me some concern when it comes to patient safety. One of the first steps with your medical device is to identify how your product can cause harm. Then you develop “controls” to prevent or mitigate the risk of harm from occurring. This process is done collaboratively with experts in different areas (software, medical, risk management. quality, etc).
In my experience, the process can take a large amount of time and requires guidance. To help understand what you need to do and how to do it, there is a standard called ISO 14971. The standard defines risk as the product of the probability of occurrence and the severity of the harm. The matrix below in an example of how our teams have measured and managed risk on several projects.
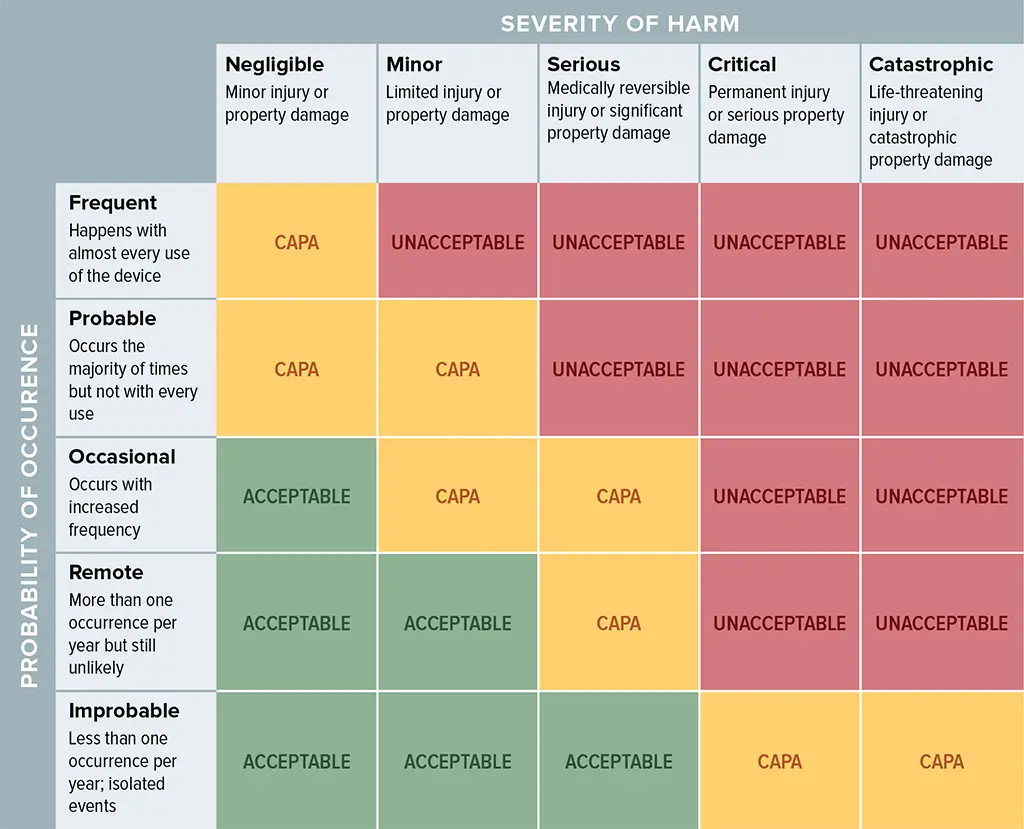
Let’s work through an example of a risk and how we develop controls to improve safety. As example, let’s say our medical device had a risk of:
Delivering a higher dosage of medication than intended could lead to patient harm
We then look at the probability of that event occurring. This can require a lot of brainstorming and research into how said event could occur. Next, we also look at the severity of the risk occurring. Using the table above, we can tell if our risk falls into one of three categories:
- Acceptable (green) -the risk is acceptable and no other risk controls are required.
- Unacceptable (red) – the risk is unacceptable and we must develop controls that either reduce the probability of occurrence and/or severity of the harm
- CAPA (yellow) – in this category additional research is needed to decide what to do. The safest thing to do is to provide additional controls to mitigate the risks.
We are problems solvers and want to help you with your next project. For a limited time, we are offering free consultations
Implement a change management process
One of biggest issues in developing medical device software is change management. Through research performed using the MAUDE database, the FDA has found that changes to software after it has already been released are where a majority of safety issues arise. You have to analyze all aspects of a change when doing your next release. This is often referred to as fit, form and finish (FFF).
Agile methodology has become more prominent in the development of medical device software. According to TIR45:2023 from the Association for the Advancement of Medical Instrumentation (AAMI), some of the benefits agile provides are:
- faster time to market
- improved quality through iterative development
- adaptation to changing requirements
- Choose a software tool to track all change requests. Tools like Jira, Azure DevOps, and GitHub can help with this.
- Include the right team members in your change control board. This may include verification, marketing, project managers, software engineers, hardware engineers, etc.
- Trace matrixes are a great idea so that you understand the full impact of a change. A simple software change may have more of an upstream or downstream impact than you think.
- Test the change, perform adequate regression testing and document the results of the testing. This is vital for FDA audits.
Understand the regulatory requirements
More and more medical devices are being connected to the Internet which opens the possibility of cyberattacks. In March of 2023, the FDA added Section 524B to the Federal Food, Drug and Cosmetic Act as a result of the Omnibus Appropriations Bill. This means that all new medical devices submitted for FDA approval require a plan outlining how they will actively identify, monitor and address potential cybersecurity threats.
Another regulatory requirement that LFT Health has helped our customers with is the IEC 62304 standard. The standard helps manufacturers by breaking the software down into three levels of concern (A, B and C). A has the least amount of concern and therefore less documentation and rigor, while C components can cause series injury or death.
We have used this approach on several medical device projects to reduce cost and time to market without sacrificing regulatory compliance and safety. To accomplish this, we do the upfront work in the design phase to make sure our C components are isolated and then properly tested.
Test. Then test some more
I can’t stress this last point enough. You need an independent verification and validation (V&V) group who is not involved with the design and implementation of the software. The FDA expects this for your development process. What is an independent verification/valid group? Their job is to test that the software meets the requirements; they are also not involved in the design and implementation of the software. The reason that they are independent is due to bias that the software engineers have. For example, when an engineer reads a requirement and implements it, they assume it is correct. However, someone who is independent may look at the requirement different. Testers who are independent will also use the product differently than the engineers and find things that the engineers didn’t even consider.
I have often had verification and validation explained to me this way. Verification is did we build the product right? In other words, does the product mean the requirements. Validation is did we build the right product? It is subtle but important difference. When we validate, we want to make sure we achieve the outcomes and that our customers’ needs are met.